|
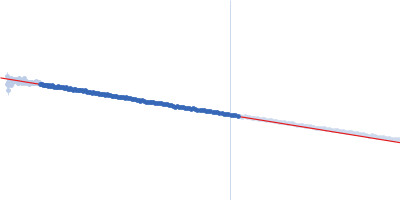
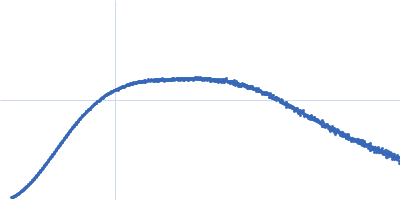
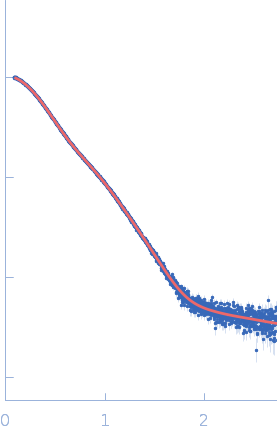
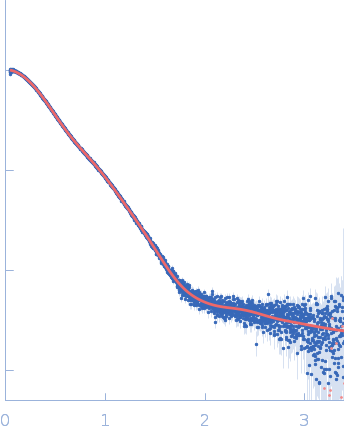
Synchrotron SAXS data from PBP5 in 100 mM Tris·HCl at pH 8.0, 150 mM NaCl, 1mM EDTA and 1mM DTT was collected on the B21-DLS beam-line at the Diamond Light Source (Didcot, UK) using a Pilatus 2M detector at a sample-detector distance of 3.7 m and at a wavelength of λ = 0.094 nm (I(s) vs s, where s = 4πsinθ/λ, and 2θ is the scattering angle). In-line size-exclusion chromatography (SEC) SAS was employed using an Agilent 1200 HPLC System by injecting a 70 μl sample at 12 mg/ml under a flow rate of 0.16 ml/min onto Shodex KW403-4F column (Showa Denko KK) at 10°C collecting 885 successive two-second frames. The scattering intensities from PBP5 elution peak region (26-frames), in addition to selected buffer scattering regions, were were azimuthally averaged, and the buffer scattering subtracted, using the ScÅtter software (version 3Q) to produce the averaged SEC-SAXS profile. From this profile, the pair-wise distance distribution function, P(r), was obtained by indirect inverse Fourier Transform with GNOM (version 5.0) using a momentum transfer range of 0.0845 < s < 3.1817 nm-1. The Rg values were estimated by applying the Guinier approximation in the range s < 1.3/Rg. The displayed SAXS-generated ab initio molecular reconstruction of PBP5 was obtained using DAMMIF (version 3.0, from ATSAS) - DAMFILT occupancy and volume-corrected bead model - by clustering and averaging dummy residue models from 20 independent runs, with an averaged Normalized Spatial Discrepancy (NSD) of 0.628.
We used AlphaFold2 (Jumper et al., 2021) to predict the structure of PB5P40-685. We did not use template structures in the prediction, iterated for up to 48 recycles, followed by energy refinement with AMBER using default settings implemented in ColabFold (Mirdita et al., 2022) and using MMseqs2 for creating multiple sequence alignments (Steinegger and Söding, 2017). The modeling confidence was assessed by the pLDDT metric and the predicted alignment error (PAE), i.e., the uncertainty about the interface. Values of pLDDT > 90 are expected to have high accuracy. Since PB5P was purified as a His-tagged fusion protein, we modeled the extra amino acids at the N-terminus with EOM (Tria et al., 2015). We then performed SAXS refinement through flexibility (SREFLEX) of the model by sampling possible conformational changes that improve the agreement with the experimental SAS profile using Normal Mode Analysis (Panjkovich and Svergun, 2016).
|
|
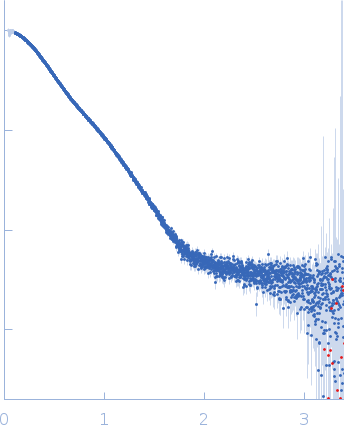 s, nm-1
s, nm-1