|
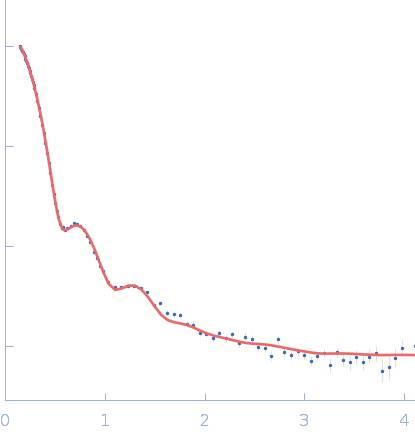
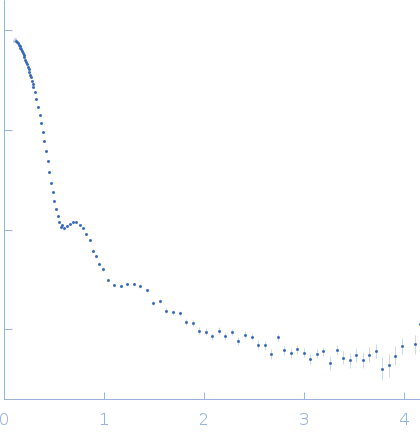
SAXS data from a solution of human methylamine-treated glycosylated alpha-2-macroglobulin (molecular mass of roughly 720 kDa for the tetramer of 180 kDa subunits) in a 20 mM HEPES, 150 mM NaCl, pH 7.4, buffer were collected at the in-house Bruker AXS NanoSTAR instrument with an Excillum Ga Metal Jet X-ray source at Aarhus University (I(s) vs s, where s = 4π sin θ/λ; 2θ is the scattering angle; λ = 0.134 nm). The instrument has a homebuilt scatterless pinhole in front of the sample and is equipped with a Bruker AXS Vantec2000 detector. Data were collected for 1800 s for the sample and buffer, respectively, and the data were normalized to absolute scale using the scattering from pure water. The structure was modelled using home-written software, which performs rigid body refinement on the structure with P222 symmetry and takes into account a hydration layer.
|
|
 s, nm-1
s, nm-1